氢溢流是一种典型的界面动态行为,是指在固体表面氢富集区(“活性位”)吸附或形成的活性氢原子在相同条件下动态迁移到另一个不吸附或不形成氢物种的氢匮乏区(“惰性”载体)。这种现象普遍存在于各种化学过程中,如催化加氢和储氢。目前,氢溢流已通过设计单原子合金(SAAs)催化剂得到有效利用,这些催化剂通常由金属载体(如Cu、Ag和Au)与原子分散的铂族金属(PGM,如Pd和Pt)组成。先前的实验观测和理论计算均表明,SAAs的活性位点可能会因为H吸附而失活,这将会阻碍持续的H2活化进程。因此,理解促进氢溢流的驱动力以及在原子尺度下洞察持续性的H2解离和H溢流过程变得至关重要。
近日,我院林森教授课题组基于密度泛函理论(DFT)数据,通过机器学习加速的分子动力学模拟,探讨了H2气体氛围下Pt/Cu(111)单原子合金表面的H溢流机理。研究发现,当H2在Pt活性位点解离时,Pt原子会由于H原子吸附而失活。有趣的是,H2与吸附的H原子之间的碰撞可以促进H溢流,使Pt原子恢复活性,从而实现可持续的H溢流过程,如图1所示。该工作开创性地阐明了气体分子与表面吸附物之间的相互作用作为一种驱动力,在气体协助的多相催化反应中发挥着至关重要的作用。
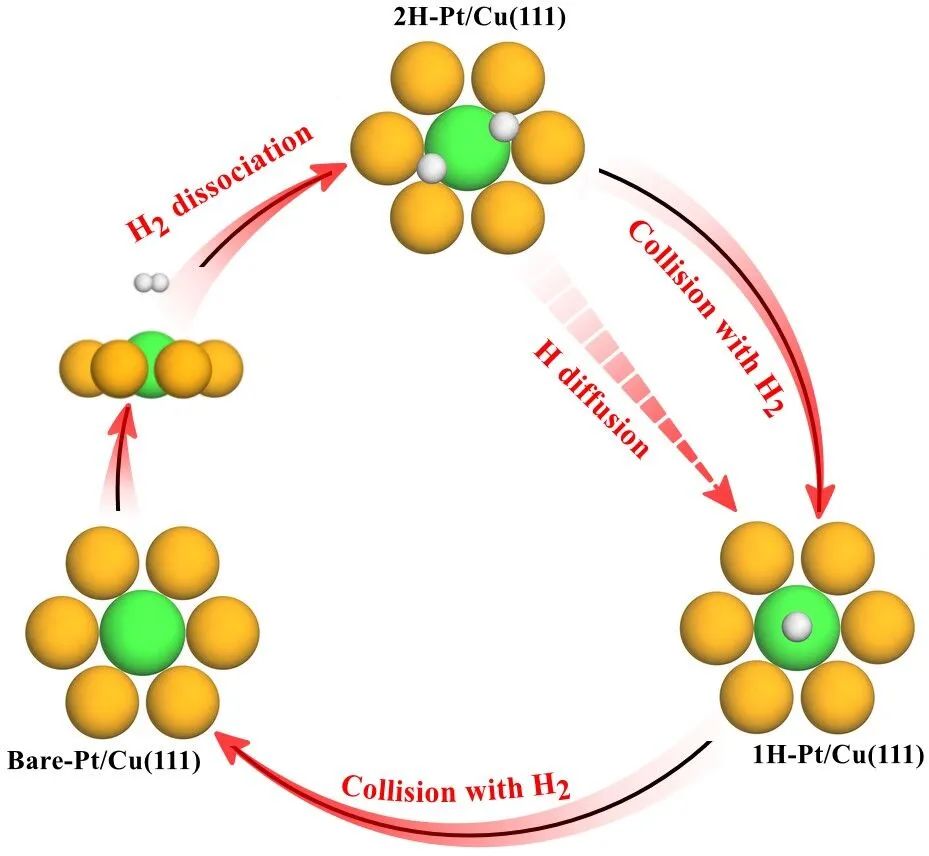
图1. H2解离和H溢流过程的示意图
在另一项工作中,课题组重点关注烷烃在金属催化剂上的活化问题,其涉及一种前驱体介导的机制,其中烷烃分子首先在催化剂表面被捕获,形成吸附的前驱体,然后在催化剂表面进行广泛扩散,以寻找活性位点。这种机制的一个特征是,在低入射能下,初始粘附概率(S0)会随着入射能的减小而增加。这种"负活化"在重构的具有缺失行结构的Pt(110)-(2×1)表面(图2)上已经被实验观察到。基于第一性原理训练数据集,课题组使用机器学习构建范德华校正的高维势能面,对Pt(110)-(2×1)上的甲烷解离进行了深入的理论研究。通过势能面计算了准经典轨迹,以模拟不同入射能下CH4和CHD3的解离。结果表明,与之前的理论研究相比,在高入射能下,初始粘附概率与实测值之间的吻合度显著提高,这表明解离势垒得到了更好的描述。此外,在低入射能下,俘获概率随着入射能的降低而增加,这与实验中观察到的低于10 kJ/mol的"负活化"相一致。这些模拟为理解烷烃异相催化过程中初始(通常是限速步骤)步骤的动力学提供了重要理论见解。
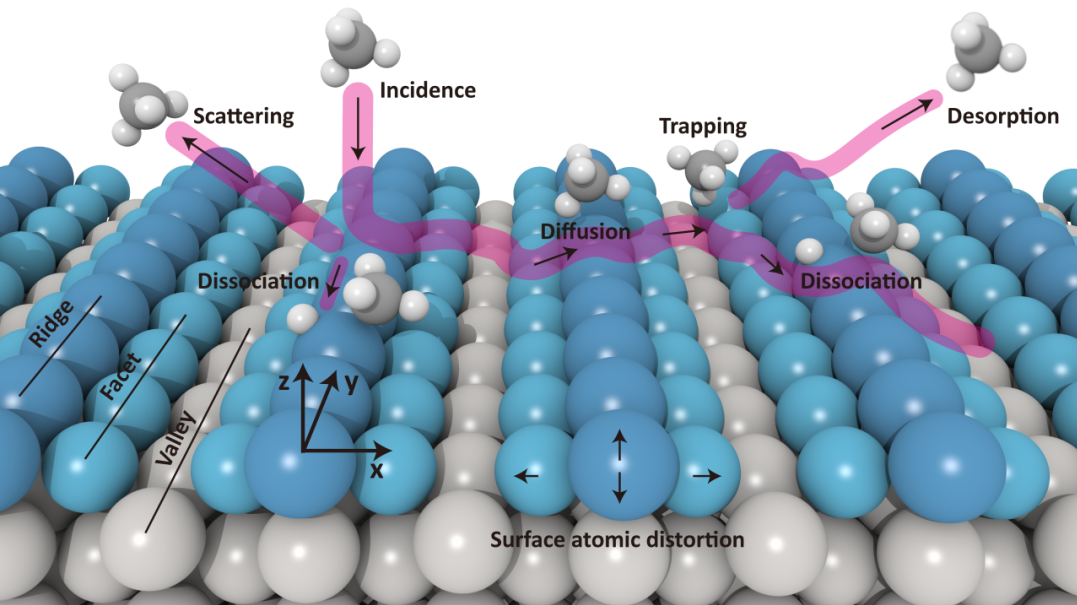
图2. 甲烷在 Pt(110)-(2×1)表面的散射、捕获、扩散、解离和吸附示意图。
成果分别以“Sustained Hydrogen Spillover on Pt/Cu(111) Single-Atom Alloy: Dynamic Insights into Gas-Induced Chemical Processes”和“Direct or Precursor-Mediated? Mechanisms for Methane Dissociation on Pt(110)-(2×1) at Both Low and High Incidence Energies”为题发表在Angew. Chem. Int. Ed.和JACS Au上,我院博士研究生顾凯旋和卫奋飞分别为第一作者,林森教授为通讯作者。相关研究得到国家自然科学基金面上项目、福建省“雏鹰计划”青年拔尖人才计划、福建省重点项目等经费资助。
文章链接:
https://doi.org/10.1002/anie.202312796
https://doi.org/10.1021/jacsau.3c00387